

Launching a successful Software as a Medical Device (SaMD) requires more than great code and innovative features, it requires a well-defined regulatory strategy. In my work, I often meet passionate founders and visionaries whose innovations face significant delays in market entry due to regulatory challenges — challenges that a clear regulatory strategy for medical devices can prevent. Too often, compliance is treated as an afterthought rather than a necessary component of success. In this article, I would like to share key perspectives that are crucial when defining a robust regulatory strategy for your product. When approached thoughtfully, regulatory planning not only helps you avoid costly mistakes and delays, but also enables you to fully realise your product’s potential, as well as minimise the maintenance cost post-market.
Regulatory strategy is a structured plan that outlines how a product will meet regulatory requirements to achieve approval and maintain compliance throughout its lifecycle. Before we dig into details, let’s look at the end-to-end process of regulatory strategy definition.

1. Define Intended Purpose of your product
This is a seemingly obvious first step to do, but many times I see people assuming that they know the intended purpose and avoid putting it into writing. It is a big mistake, as many times the way you phrase your intended purpose will severely impact your product regulatory status and its potential impact on patients’ safety, as a result changing the regulatory expectations and required clinical/performance evidence.
Each market regulator has their own definition of the intended purpose, or slightly different terminology, while the overall concept remains the same. As an example, intended purpose is defined by Article 2 of EU MDR as:
‘intended purpose’ means the use for which a device is intended according to the data supplied by the manufacturer on the label, in the instructions for use or in promotional or sales materials or statements and as specified by the manufacturer in the clinical evaluation;
Intended purpose is a cornerstone document describing your medical device. It shall be defined early, and should be used as an input to several other product lifecycle activities, including regulatory strategy, risk assessment, product requirements definition, clinical evaluation, usability studies, and so on. Key aspects to be covered by the intended purpose:
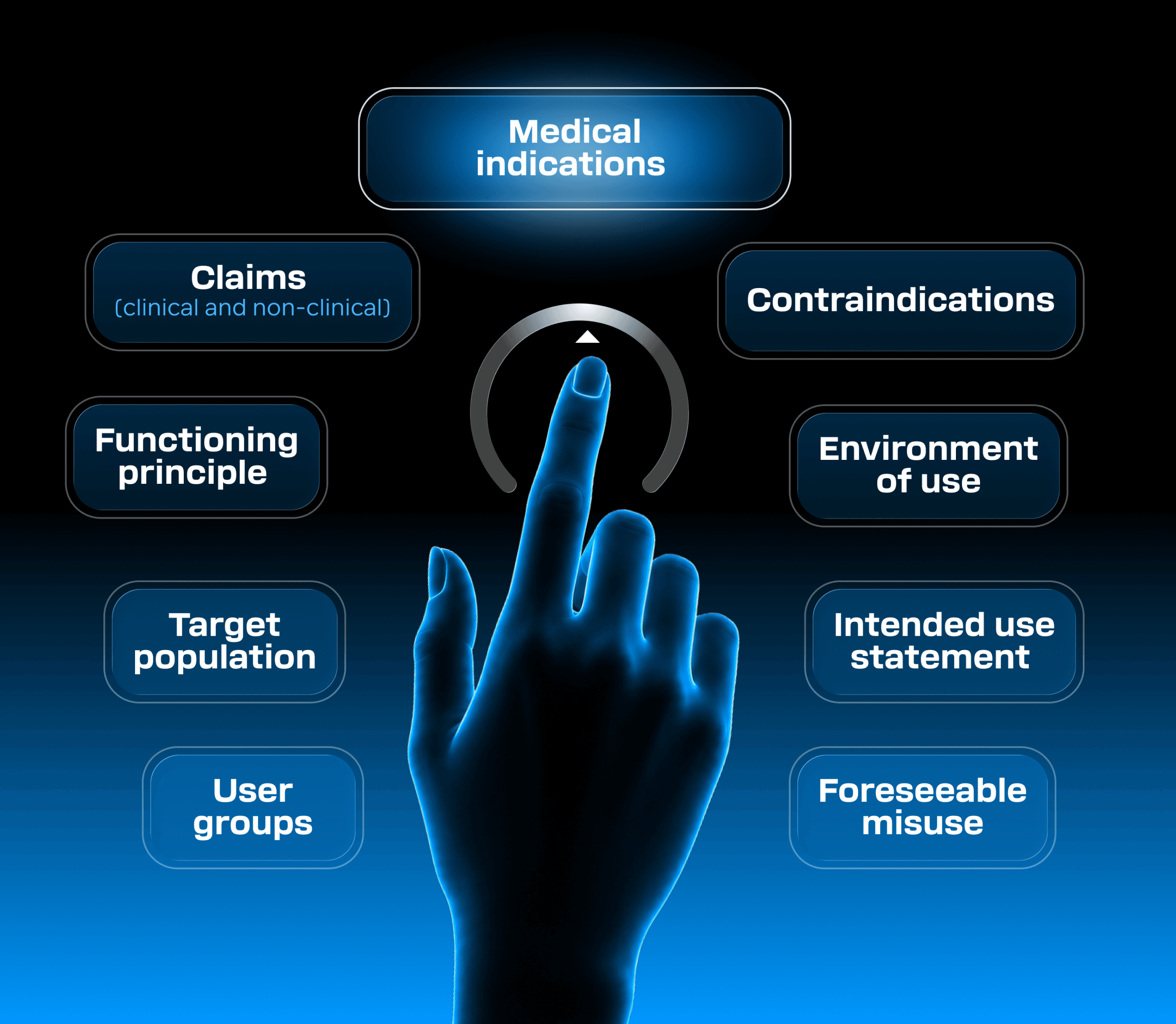
When defining the intended purpose of your product, it’s important to strike the right balance. You need to be precise enough to support your claims with appropriate clinical evidence, while still leaving room to evolve the product within regulatory boundaries.
2. Define your business and product priorities
To define an effective regulatory strategy for your organization, you first need to be clear on your main business goals. Are you aiming to enter a specific market as quickly as possible, regardless of cost? Or is the priority to get the product into any market to support discussions with investors? Perhaps the focus is on optimizing market entry from a budget perspective. Each of these drivers plays an important role and should guide how you shape your regulatory approach.
It’s equally important to think about your long-term product vision. How will your product fit into the existing healthcare ecosystem? Will it need to integrate with third-party tools? Will it require frequent updates after launch? Or are you building a platform that will expand to include multiple medical devices over time?
Considering these factors early on helps you position your product appropriately from a regulatory standpoint. Thinking about the end goal from the start allows you to choose a path that minimizes friction, avoids unnecessary rework, and supports your broader strategy.
Finally, from a business perspective, it is worth considering an evolutionary approach: assess whether there is a way to define your product's intended purpose so that it falls outside the medical device scope entirely, or within a lower classification. This can allow you to place an initial version of the product on the market earlier, while you work on the full-scope version of the product, which may require more time to obtain approval.
3. Prioritise markets of interest
Once you’ve clarified your business goals and long-term product vision, the next step is to define a prioritized list of target markets. It might be tempting to focus on just one market, but thinking more broadly early on can make a big difference.
The reason is simple: optimization. Knowing where you’re heading from the start helps you make smarter operational decisions. It allows you to reuse work across different markets, run certain activities in parallel, and take advantage of early feedback from regulators or other stakeholders.
For example, your immediate priority might be entering the European market. However, engaging early with the FDA through a Q-submission meeting could provide valuable insights if you’re planning to expand into the US in the near future.
While maintaining focus is important, having a broader perspective is just as critical. It helps you build a more efficient execution strategy and can save you significant time, cost, and frustration down the line.
4. Perform product qualification and classification
The next step is to determine the regulatory status of your product. At this stage, the key question is whether your product falls within the scope of a specific regulation; this is known as product qualification.
To do this, you need to review the relevant definitions within the applicable regulatory framework and assess whether your product, as described by its intended purpose, fits within any of them. For medical devices, one of the first and most important questions is whether your product meets the definition of a “medical device.”
In addition to the regulations themselves, most regulatory authorities publish guidance documents that help clarify how these definitions should be interpreted. These resources are essential and should be carefully considered during the product qualification process.
Once you’ve established that your product falls under a specific regulation, the next step is to determine its classification. Medical devices are grouped into different classes based on their potential impact on patient safety. The classification rules and the classes themselves vary across markets.
To classify your product, you need to follow the applicable regulatory guidelines for your target market and assign it to the appropriate class. It’s important to clearly document the rationale behind your classification decision. While it is the manufacturer’s responsibility to classify the product, the boundaries between classes can sometimes be unclear. A well-documented justification can help prevent complications later on.
In some cases, both product qualification and classification can be complex or ambiguous. In such situations, it may be necessary to seek input from regulatory authorities or other relevant stakeholders. Having a clearly defined intended purpose and an initial regulatory strategy will support these discussions and help others assess and provide feedback on your approach.
Once you have determined your product classification, it is worth taking the time to understand the applicable regulatory requirements, guidelines, common specifications, and harmonised or recognised standards. For software medical devices, some of the foundational standards to be aware of include ISO 13485, IEC 62304, IEC 82304, IEC 62366, ISO 14971, and ISO 15223-1.

5. Review your product's high-level architecture
The next step is to examine your product architecture closely, particularly if your product is a software medical device. Most regulators allow you to draw a clear boundary between medical and non-medical components. As long as a non-medical component does not contribute to the device's intended purpose or serve a safety function, it can be excluded from the medical device scope. This can significantly simplify post-market maintenance and change control, especially if you anticipate frequent updates to the non-medical parts. That said, non-medical components must not affect how the device performs its intended function, and a clear interface should be defined to separate the two. It's worth noting that qualification criteria differ significantly across markets, so if you operate globally, factor this in early.
A well-defined product architecture is also the foundation for IEC 62304 compliance. The standard is considered state-of-the-art for medical device software and requires you to assign a safety classification to each software unit. The justification for each classification, along with the qualification of each unit as medical or non-medical, must be clearly documented in the technical documentation.
As the product evolves, a non-medical component can drift into medical territory. It is worth implementing a change management process and regularly reassessing the qualification and boundary of the medical device scope to avoid potential non-compliance.
It is also worth considering cybersecurity as part of your architecture review. Even if certain components are excluded from the medical device scope, those that connect to or interact with the medical portion of your product can represent a cybersecurity attack surface. Regulators, particularly FDA, increasingly treat cybersecurity as a crucial requirement and expect manufacturers to address it explicitly in their technical documentation, regardless of how the device boundary is drawn.
6. Define clinical evidence strategy
Clinical evidence strategy defines how you will demonstrate your product's safety and performance. The approach depends heavily on product classification, intended purpose, technology, available clinical data for similar products, and market-specific regulatory requirements. All of these factors must be considered together to define a strategy that enables the business rather than slows it down.
Product risk classification and novelty are the two primary drivers of your clinical evidence approach. Some regulations, such as EU MDR/IVDR, explicitly state that clinical investigation data is required for higher-risk devices, while accepting a literature-based clinical evaluation for lower-risk classes. Establishing this early helps you avoid costly rework and delays.
A formal, documented clinical evaluation is required in some markets even for the lowest device classes. For most devices, it begins with a systematic literature review to explore the state of the art, comparable devices, and available clinical data. Once complete, the critical question is whether this data is sufficient to demonstrate the device's safety and performance, including scientific validity, analytical performance, and clinical performance. Where gaps exist, you will need to plan how to address them, whether through retrospective studies drawing on existing clinical data or prospective clinical investigations. Most regulators have specific requirements governing how clinical data may be collected; these must be factored in early.
Across markets, regulators allow you to leverage clinical data from an already-marketed equivalent device rather than generating entirely new evidence. In the United States, the 510(k) pathway is based on demonstrating substantial equivalence in terms of intended use and technological characteristics. In markets such as the EU, the bar is higher, as demonstrating equivalence under EU MDR requires evidence across clinical, technical, and biological dimensions, and where the equivalent device is not your own, you may need contractual access to its technical documentation.
Even after a product has been successfully placed on the market, it is important to plan upfront how you will sustain the evidence base over time. Post-market surveillance and post-market clinical follow-up (PMCF) activities serve this purpose, enabling you to generate additional clinical evidence and respond to emerging signals throughout the product's lifecycle.
Clinical evidence requirements differ meaningfully across markets. A study designed for one jurisdiction may not satisfy another. Where possible, design clinical investigations to generate data reusable across markets, reducing duplication of effort.
As a closing note, it is worth highlighting that clinical evidence feeds directly into the benefit-risk determination, which is a cornerstone of conformity assessment in most markets.
7. Prepare a compliance roadmap
Once all of the above has been addressed, you are ready to build your compliance roadmap, mapping all required activities in a logical sequence with defined timelines. A compliance roadmap is not a static document, it is a living plan that evolves alongside your product and should be revisited regularly as your understanding of regulatory requirements matures.
Quality management system. The foundation of your compliance roadmap is the establishment and implementation of a quality management system (QMS) aligned with ISO 13485 and relevant market regulatory requirements. The QMS must be in place before most other regulated activities can formally begin, as it defines the processes under which your product is designed, developed, and maintained. For early-stage companies, it is important to size the QMS appropriately, as overly complex systems create unnecessary overhead, while under-defined systems create compliance gaps that are costly to fix later.
Medical device documentation and design controls. Closely linked to the QMS is the establishment of design controls and the product technical documentation. Design controls ensure that your product's development is traceable from user needs through to verified and validated outputs, and that changes are managed in a controlled way. For software medical devices, this includes maintaining a software development lifecycle aligned with IEC 62304, covering risk management (safety, usability, cybersecurity, technological risks), requirements, architecture, detailed design, implementation, verification, and problem resolution. Usability engineering, governed by IEC 62366, is often underestimated but is a core part of the development process. It ensures your device works the way real users expect it to, and that the design has been tested and validated to confirm that use-related risks are effectively controlled. Technical documentation must demonstrate conformity with all applicable regulatory requirements and is the primary artefact reviewed during conformity assessment.
Regulatory interactions and external dependencies. Plan your interactions with regulators and other regulatory actors early and build them explicitly into your timeline. For products requiring notified body involvement in Europe, be aware that notified body capacity has been significantly constrained since EU MDR came into full effect, and securing a notified body agreement can take considerably longer than expected (conformity assessment procedure may take up to 12-24 months for some devices). Similarly, if you plan to engage with FDA, Q-submission meetings should be scheduled well in advance, as they provide valuable early alignment and can prevent costly pivots later. Other regulatory actors to account for include authorised representatives, importers, and distributors, each of whom has defined responsibilities under applicable regulations.
Post-market surveillance. Compliance does not end at market entry. Your roadmap should include a post-market surveillance system from the outset, covering how you will collect, analyse, and act on real-world data about your device's safety and performance. In addition, it is important to monitor the evolving regulatory landscape, as requirements across markets continue to develop and change.
8. Other aspects to consider
While this article has covered several key aspects of regulatory strategy for medical device software, it is important to highlight that documenting a clear regulatory strategy is valuable even for products that sit just outside the medical device scope. Regulatory boundaries are not always permanent, and having a strategy in place early ensures you are prepared when they shift.
As a final note, it is worth emphasising that defining your regulatory strategy early allows you to avoid costly mistakes, ensures a streamlined product lifecycle and evidence collection, and ultimately brings your innovation to market safely and efficiently.
For this reason, engaging with experienced medical device regulatory consulting experts early in the product journey can make a significant difference. At Star, we help founders and MedTech teams get critical decisions right from the start — from defining the intended purpose and product classification to developing a clinical evidence strategy that supports the chosen regulatory pathway. By establishing a strong regulatory foundation early, we help companies create a roadmap that reduces uncertainty, avoids costly rework, and supports successful market access as their products evolve.






